Proteomics > Which service should I request? > Proteome Identification and Quantification > Low-input analysis
When limited sample amount is availible, optimal sample preparation and sensitivity for a good proteome depth is required. Especially for the sample handling, large surfaces and sample transfer steps need to be avoided. Moreover, for a sucessfull enzymatic digestion, reaction volume has to be small.
We divide the low input samples in two categories but it is important to keep in mind that based on the cell type, the protein amount varies.
In case you want to analyze a subpopulation of cells from FACS sorted cells and you are limited with the number of cells you can provide us for the Proteomics analysis, we have implemented a workflow for low-input samples from FACS sorted cells (<5k cells/sample).
Below you find more information. But briefly, we have seen that 3000-5000 cells/replicate are ideal. Sorting less cells will decrease the protein identification and in case of more cells, we risk to have cross contamination in the collection plate because the wells are too small.
For the suggested workflow we use Eppendorf twin.tec® PCR Plate 384.
 Consider:
Consider:
Some important points:
Low-input analysis
Table of contents
1.General description
When limited sample amount is availible, optimal sample preparation and sensitivity for a good proteome depth is required. Especially for the sample handling, large surfaces and sample transfer steps need to be avoided. Moreover, for a sucessfull enzymatic digestion, reaction volume has to be small.
We divide the low input samples in two categories but it is important to keep in mind that based on the cell type, the protein amount varies.
< 50k cells/sample
Dependent on how many cells you can collect, the sample preparation is either done in an Eppendorf tube or in 384 well plates. The LC-MS analysis will be performed on an Evosep coupled to a TIMS-TOF Pro or with our standard LC-MS setup with an waters Mclass nanoLC coupled to an Thermo Orbitrap::Iontrap mass analyzer.In case you want to analyze a subpopulation of cells from FACS sorted cells and you are limited with the number of cells you can provide us for the Proteomics analysis, we have implemented a workflow for low-input samples from FACS sorted cells (<5k cells/sample).
Below you find more information. But briefly, we have seen that 3000-5000 cells/replicate are ideal. Sorting less cells will decrease the protein identification and in case of more cells, we risk to have cross contamination in the collection plate because the wells are too small.
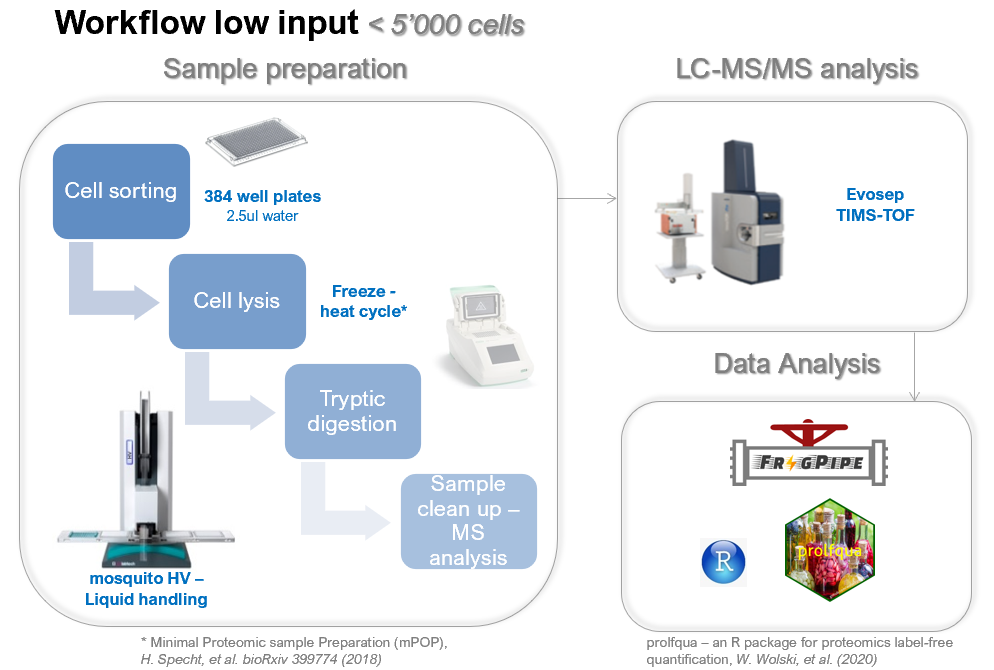
Sample sorting (< 5k cells)
Ideally you sort your cells in a low volume of water (our standard is 2.5ul), followed by snap freezing your cells and storing at -80°C.For the suggested workflow we use Eppendorf twin.tec® PCR Plate 384.
- The FACS sorting buffer should not contain any FBS or other protein contaminations.
- The final volume after collecting should be as small as possible. Cells need to be sorted in single cell mode.
- To avoid evaporation during sample preparation, don't sort in the border wells of the 384 well plate (A1-> A24, A1->P1, A24->P24 and P1-> P24).
- Sort your replicates per column | and not per line!
- If you wish a label free quantification, you must provide us at least 5 samples per condition.
Workflow
You can also obtain more information in the following publication of Daniel Gonzalez-Bohorquez et al. for our current low-input workflow:Protein identifications
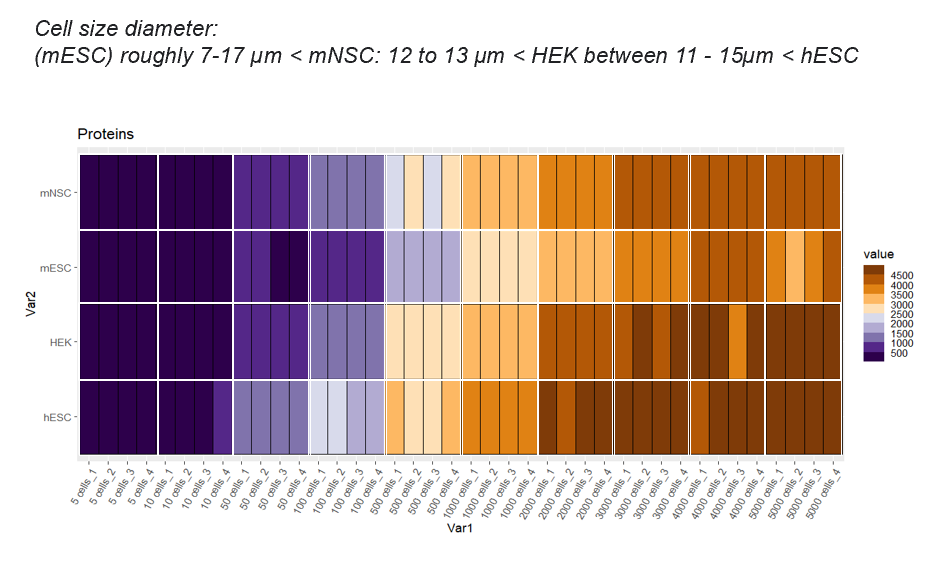
The number of identified proteins depends on the cell type and the cell number. Here some examples of cell titrations/replicates and what we can achieve :2. Protein digestion
The first step for the identification of unknown proteins is protein digestion using proteases (e.g. trypsin). We can process samples in the following forms:| # | Examples of digestion protocols | Comments |
| 384 well plates | in solution | IMPORTANT, sort the cells in single-cell mode in a 384-well plate ideally in water and snap freeze the plate directly. Whenever possible, use volatile buffers. Limit the amount of SDS and avoid CHAPS/Tween. |
| 1.5ml Eppendorf | Urea based digestion | More cells are required (> 50'000) for a higher volume in-solution digestion |
| Laser microdissection from tissues | in solution protocol | see LMD Proteomics. |
3. LC-MS analysis
At FGCZ, the default LC-MS approach for the low input samples is based on:- liquid chromatography separation on the Evosep One coupled
- data acquisition on the TIMS TOF systems (Brukerusing either Data Dependent Acquisition (DDA) or Data Independent Acquistion (DIA) with Parallel Accumulation and Serial Fragmentation (PASEF).
4. Data analysis
For the identification and quantification of proteins, the information within the acquired raw files is extracted and matched against protein FASTA databases using a search engine.Some important points:
- if a protein is not in a database, it cannot be identified using the standard approaches
- for common organisms (e.g., human, mouse, ...) the databases are well-annotated and reviewed. We mainly use Uniprot databases.
- As search engine for the low input workflow we use either FragPipe for the database search (ddaPASEF) or the lable free approach from DIANN (diaPASEF)
- Data analysis using linear or mixed statistical models, using our published prolfqua package.
- Generation of reports, tables and plots
- NEW (Oct. 2023): visualization of data in exploreDEG Interactive Shiny App!
- (for selected species, e.g. human and mouse, Gene Set Enrichment Analysis (GSEA) and Over-Representation Analysis (ORA) are performed using our fgcz.gsea.ora package, which integrates WebGestalt)