Proteomics > Which service should I request? > Glycan / Glycoprotein analysis > Identification of glycoproteins
Questions?
The identification of the candidate glycoproteins is performed using a standard LC-MS/MS Protein identification workflow. The approach does not focus on glycosylated peptides and the protein identity is obtained by observing only the peptides that do not undergo glycosylation.
The approach can be used in combination with quantitative strategies.
Depending on the type of samples:
Examples of instruments are the Q-Exactive (Thermo) or, in case of experiments that require higher sensitivity, Orbitrap Lumos(Thermo)
See all out instruments in our (Instrument Intranet page)
The proteins including the N-X-S/T (X≠P) are considered as potential glycoproteins.
Identification of glycoproteins
Table of contents
General description
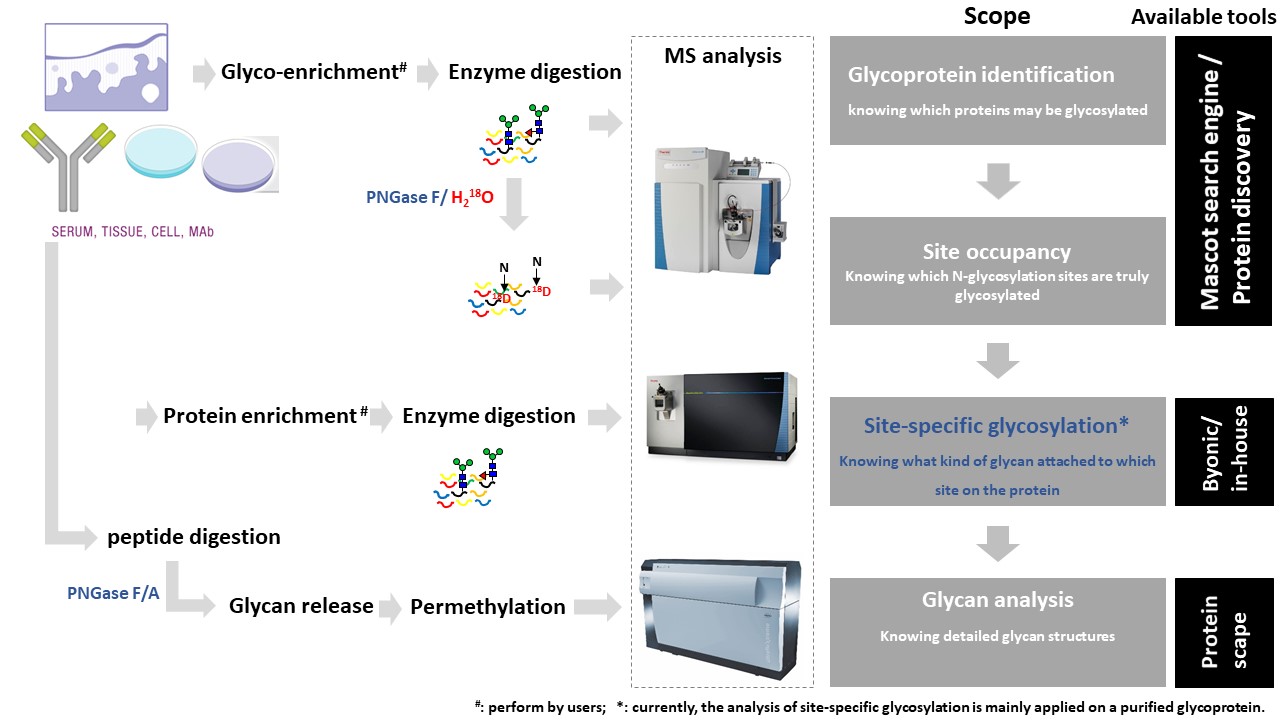
The scope of glycoprotein identification is to obtain a list of potential glycoprotein candidates starting from a complex protein extract. The workflow is based on the fact that N-glycosylation occurs at asparagine residues in the consensus sequence N-X-S/T (X≠P).Questions?
- Check our FAQs Glycan / Glycopeptide analysis page or contact us at proteomics at fgcz.ethz.ch
Workflow
Based on the biological question and on the type of sample, a typical workflow for processing the samples prior to MS and data analyses consists of:The identification of the candidate glycoproteins is performed using a standard LC-MS/MS Protein identification workflow. The approach does not focus on glycosylated peptides and the protein identity is obtained by observing only the peptides that do not undergo glycosylation.
The approach can be used in combination with quantitative strategies.
1. Glycoprotein enrichment
Enrichment of glycoprotein from a protein pool using lectins or antibodies (performed by the customer)2. Protein digestion
The first step for the identification of unknown proteins is protein digestion using proteases (e.g. trypsin). In most cases the standard protein identification workflow is appliedDepending on the type of samples:
- Protein mixtures. Minimal recommended amount: 10 µg
3. LC-MS analysis
At FGCZ, the default LC-MS approach for protein identification includes data acquisition on one of our LC-MS/MS systems using Data Dependent Acquisition (DDA - also named Shotgun proteomics).Examples of instruments are the Q-Exactive (Thermo) or, in case of experiments that require higher sensitivity, Orbitrap Lumos(Thermo)
See all out instruments in our (Instrument Intranet page)
4. Data analysis
Peptide and protein identification using search engines (e.g. Mascot) and protein sequence databases. For common organisms (e.g., human, mouse, ...) the databases are well-annotated and reviewed. We mainly use Uniprot databases.The proteins including the N-X-S/T (X≠P) are considered as potential glycoproteins.